Research grants
DeMeTeR: 2024-2028
DeMeTeR: Interpreting Diffusion Models Through Representations
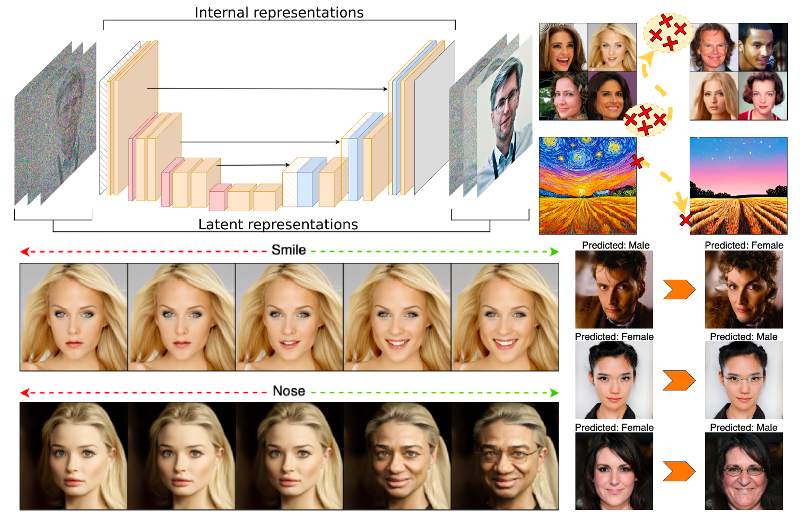
Diffusion models have been the latest revolution in the domain of generative modelling in computer vision, surpassing the capabilities of long-reigning generative adversarial networks , and are currently being adapted to multiple other domains and modalities. However, we still lack an in-depth understanding of their inner workings from both an empirical and theoretical standpoint.
Considering that, the main goals of the DeMeTeR project are:
- to broaden the practical and theoretical understanding of diffusion-specific latent representations and architecture-specific internal representations of diffusion models,
- to develop novel methods of manipulating these representations that allow for enhancing safety and explainability of deep learning models
Work on this project is financially supported by the Polish National Science Centre PRELUDIUM BIS grant 2023/50/O/ST6/00301.
PINEBERRY: 2024-2025
PINEBERRY: Explainable, Robust and Secure AI for demistyfying Space Operations
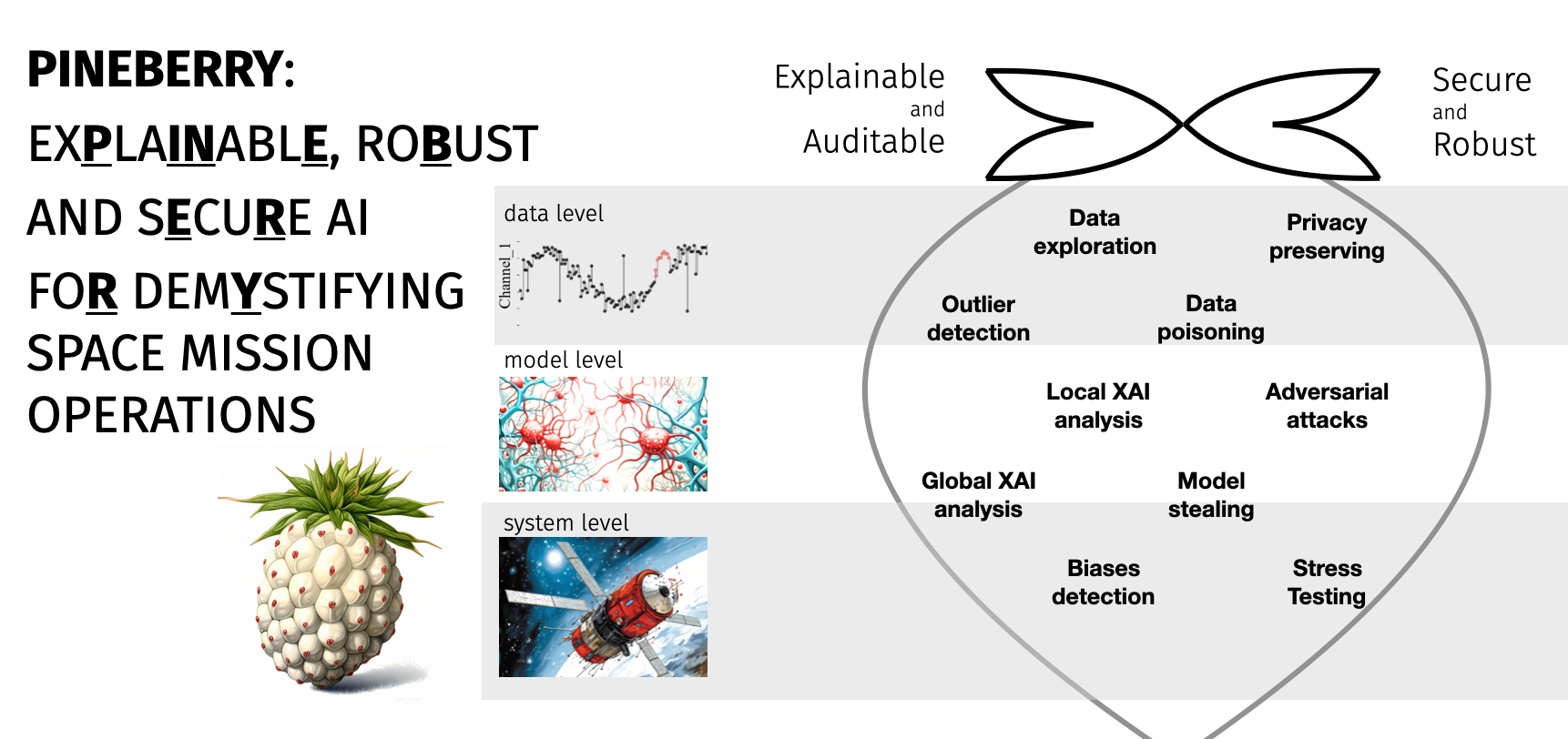
In the PINEBERRY project (ExPlaINablE, RoBust And SEcuRe AI FoR DemYstifying Space Mission Operations), we will address the important research gap of lack of “trust” into (deep) machine learning algorithms for space mission operations, through tackling real-life downstream for the A2I roadmap using deep and classic machine learning algorithms empowered by new security and explainable AI (XAI) techniques. We believe that PINEBERRY will be an important step toward not only “uncovering the magic” behind deep learning algorithms (hence building trust in them in downstream tasks), but also in showing that XAI techniques can be effectively utilized to improve such data-driven algorithms (both classic and deep machine learning-powered), ultimately leading to better algorithms. Finally, we will put special effort into (i) unbiasing the validation of existing and emerging algorithms through ensuring their full reproducibility (both at the algorithm and at the data level), and (ii) ensuring security of algorithms on different levels of data, models, and system.
To this end we develop a catalogue for issues relevant to Security and Explainability that will guide validation of ML/AI models used in SO. This project is conducted in collaboration with KP Labs company.
Work on this project is financially supported by European Space Agency grant ESA 4000144194/23/D/BL.
PvSTATEM 2023-2027
PvSTATEM: Serological testing and treatment for P. Vivax: from a cluster-randomised trial in Ethiopia and Madagascar to a mobile-technology supported intervention
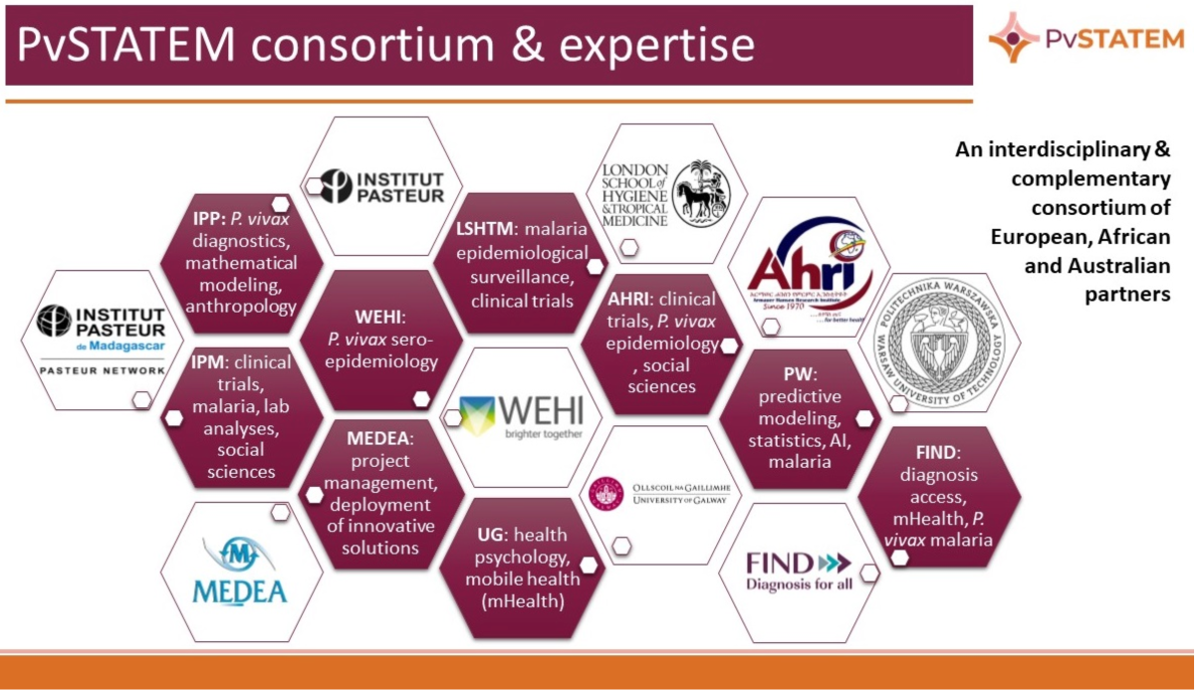
The PvSTATEM project aims to demonstrate the efficacy and the community acceptability of P. vivax Serological Testing and Treatment (PvSeroTAT), a new intervention for the control and elimination of malaria, in cluster-randomised trials in Ethiopia and Madagascar. The project will also innovate new mobile technologies for the efficient implementation of PvSeroTAT in settings beyond clinical trials. The PvSeroTAT intervention includes a serological diagnostic test that measures antibodies to multiple P. vivax antigens and informs an individual-level treatment decision. However, the results from serological tests can also inform population-level surveillance of malaria. In this Hop-on project, mathematical models, machine learning tools, and digital technologies will be developed so that data generated by the clinical trials in Ethiopia and Madagascar can inform national malaria surveillance programs.
Work on this project is financially supported by the HORIZON grant HORIZON-WIDERA-2022-ACCESS-07-01.
GliomAI 2024
GliomAI: Artificial Intelligence for Radiogenomic Atlas of Gliomas
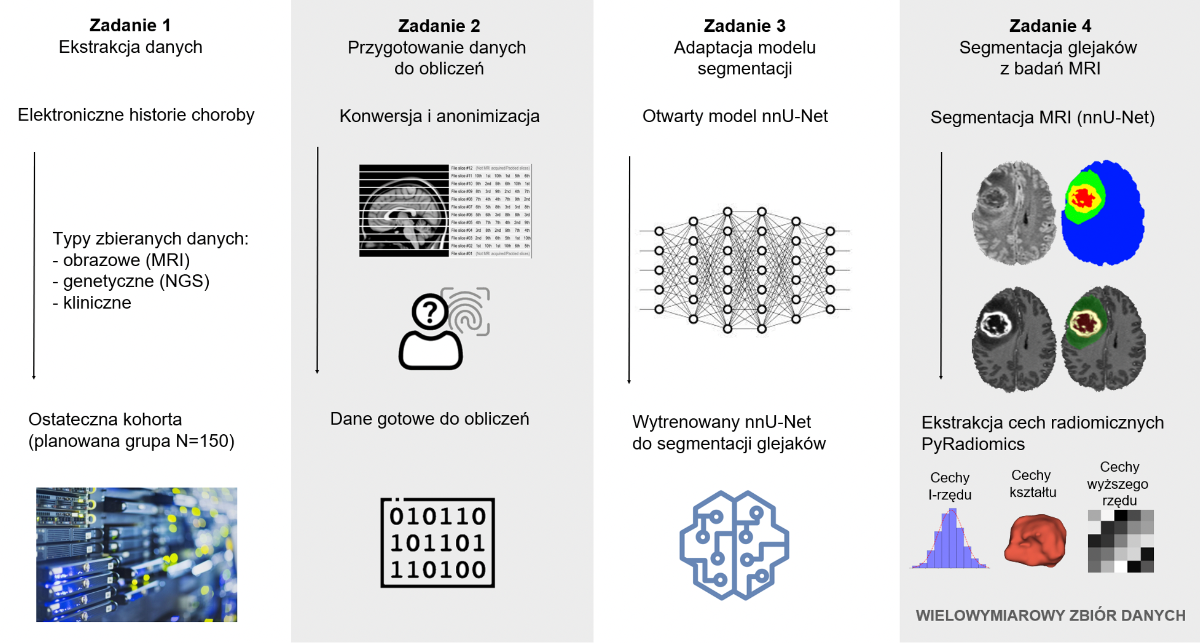
The new 2021 WHO classification of brain tumours places more emphasis than before on genetic variation in the classification of tumour lesions. However, invasive procedures are required for genetic diagnosis, which pose risks to patients and limit access to molecular profiling. Radiomics, a non-invasive approach, allows the analysis of tumour features using imaging data such as magnetic resonance imaging (MRI), which is used to extract computational independent variables. This approach allows the analysis of heterogeneity, spatial relationships and textural patterns that characterise different tumour phenotypes, however, may not be graspable by human perception. The correlation of such computational variables obtained with genetic findings is called radiogenomics.
Multidimensional datasets play a key role in the development of the field of radiogenomics. However, in order to do so, it is necessary to delineate regions of interest within imaging studies - so-called masks - which are ultimately used to extract computational variables. In this project, we plan to develop a novel radiomic database containing not only clinical, genetic and imaging data, but also the previously mentioned segmentation masks of gliomas and their immediate surroundings. To this end, an interdisciplinary research team will be formed, benefiting from the synergistic impact of the two units involved in the project at our Universities.
Work on this project is financially supported by Warsaw Medical University and Warsaw University of Technology within the Collaboration Initiative Programme WUM_PW INTEGRA 1.
PINEAPPLE 2023-2025
PINEAPPLE: Explainable AI for hyperspectral image analysis
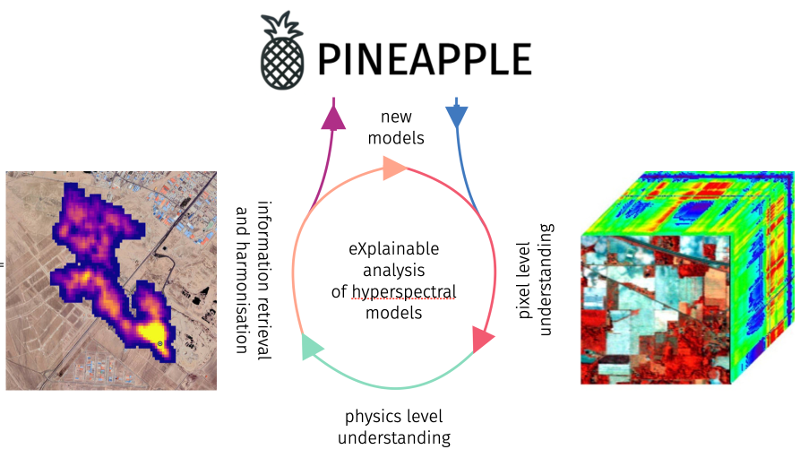
In the PINEAPPLE project (exPlaINablE Ai for hyPersPectraL imagE analysis), we will address the important research gap of lack of “trust” into (deep) machine learning algorithms for EO, through tackling two real-life EO downstream tasks (estimating soil parameters from HSI and detecting methane in such imagery) using new deep and classic machine learning algorithms empowered by new explainable AI (XAI) techniques. We believe that PINEAPPLE will be an important step toward not only “uncovering the magic” behind deep learning algorithms (hence building trust in them in EO downstream tasks), but also in showing that XAI techniques can be effectively utilized to improve such data-driven algorithms (both classic and deep machine learning-powered), ultimately leading to better algorithms.
Finally, we will put special effort into:
- unbiasing the validation of existing and emerging algorithms through ensuring their full reproducibility (both at the algorithm and at the data level), and
- understanding & improving the generalization of such algorithms when fundamentally different data is used for testing (e.g., noisy, with simulated other atmospheric conditions, captured in different area/time, and so forth)
Work on this project is financially supported by European Space Agency grant ESA AO/1-11524/22/I-DT.
ARES 2022-2026
ARES: Attack-resistant Explanations toward Secure and trustworthy AI

Machine learning explainability, fairness, robustness, and security are key elements of trustworthy Artificial Intelligence, an area of strategic importance. In this context, the main goals of the ARES project are:
- Develop adversarial attacks on state-of-the-art explanations to investigate vulnerabilities and limitations of the existing explainability and fairness approaches in machine learning.
- Introduce novel robust explanations that are stable against manipulation and intuitive to evaluate.
Achieving the first goal primarily impacts various domains of research, which currently use (and explain) black-box models for knowledge discovery and decision-making, by highlighting vulnerabilities and limitations of their explanations. Achieving the second goal impacts more the broad machine learning domain as it aims at improving state-of-the-art by introducing robust explanations toward secure and trustworthy AI.
Work on this project is financially supported by the Polish National Science Centre PRELUDIUM BIS grant 2021/43/O/ST6/00347.
DARLING 2022-2024
DARLING: Deep Analysis of Regulations with Language Inference, Network analysis and institutional Grammar
Aim of the project
Developing the tools for automated analysis of content of legal documents leveraging Natural Language Processing, that will help understand the dynamic of change in public policies and variables influencing those changes.
Those tools will be firstly used to analyse the case of development of policy subsystem regulating usage of AI in the European Union.
Specific goals of the project
- Developing and evaluating multilingual models for issue classification for legal and public policy documents.
- Developing embedding-based topic modeling methods for legal and public policy documents suited for analysis of change of the topics between documents.
- Institutional grammar based analysis of changes in topics between different public policy documents, regulations and public consultation documents.
- Agent-based models predicting diffusion of issues in public policy documents.
Methodology
The core of the DARLING project is the issues and topic analysis in documents connected with regulations development using NLP tools. Issues analysis shall allow tracking how different options of AI operationalisation, ways the AI-connected threats are perceived as well as ideas regarding AI regulations are shared among three different types of texts: scientific, expert and legal ones. The extracted issues will then be subject to complex networks analysis and institutional grammar approach. The network analysis, backed by agent-based modeling, will be used to examine the flow of issues among the documents based on their vector-formed characteristics. On the other hand, the Institutional Grammar (IG) will be used to analyze the modality of issues, e.g., the tendency to regulate a specific aspect of AI in a given issue, its deontic character or its conditionality.
In result the DARLING project will effect in the development of new methods to analyze legal documents connected to regulation based on deep text processing and links among the documents. An inter-institutional and interdisciplinary team of computer, political sciences and physics of complex systems scientists will elaborate new machine learning approaches to examine the regulation corpora, issues recognition, issues analysis by the means of IG as well as propose new methods of modeling the flow/changes of regulations based on complex networks tools.
X-LUNGS 2021-2024
X-LUNGS: Responsible Artificial Intelligence for Lung Diseases

The aim of the project is to support the process of identification of lesions visible on CT and lung x-rays. We intend to achieve this goal by building an information system based on artificial intelligence (AI) that will support the radiologist’s work by enriching the images with additional information.
The unique feature of the proposed system is a trustworthy artificial intelligence module that:
- will reduce the image analysis time needed to detect lesions,
- will make the image evaluation process more transparent,
- will provide image and textual explanations indicating the rationale behind the proposed recommendation,
- will be verified for effective collaboration with the radiologist.
Work on this project is financially supported from the INFOSTRATEG-I/0022/2021-00 grant funded by Polish National Centre for Research and Development (NCBiR).
HOMER 2020-2025
HOMER: Human Oriented autoMated machinE leaRning

One of the biggest challenges in the state-of-the-art machine learning is dealing with the complexity of predictive models. Recent techniques like deep neural networks, gradient boosting or random forests create models with thousands or even millions of parameters. This makes decisions generated by these black-box models completely opaque. Model obscurity undermines trust in model decisions, hampers model debugging, blocks model auditability, exposes models to problems with concept drift or data drift.
Recently, there has been a huge progress in the area of model interpretability, which results in the first generation of model explainers, methods for better understanding of factors that drive model decisions. Despite this progress, we are still far from methods that provide deep explanations, confronted with domain knowledge that satisfies our ,,Right to explanation’’ as listed in the General Data Protection Regulation (GDPR).
In this project I am going to significantly advance next generation of explainers for predictive models. This will be a disruptive change in the way how machine learning models are created, deployed, and maintained. Currently to much time is spend on handcrafted models produced in a tedious and laborious try-and-error process. The proposed Human-Oriented Machine Learning will focus on the true bottleneck in development of new algorithms, i.e. on model-human interfaces.
The particular directions I consider are (1) developing an uniform grammar for visual model exploration, (2) establishing a methodology for contrastive explanations that describe similarities and differences among different models, (3) advancing a methodology for non-additive model explanations, (4) creating new human-model interfaces for effective communication between models and humans, (5) introducing new techniques for training of interpretable models based on elastic surrogate black-box models, (6) rising new methods for automated auditing of fairness, biases and performance of predictive models.
Work on this project is financially supported from the SONATA BIS grant 2019/34/E/ST6/00052 funded by Polish National Science Centre (NCN).
DeCoviD 2020-2022
DeCoviD: Detection of Covid-19 related markers of pulmonary changes using Deep Neural Networks models supported by eXplainable Artificial Intelligence and Cognitive Compressed Sensing
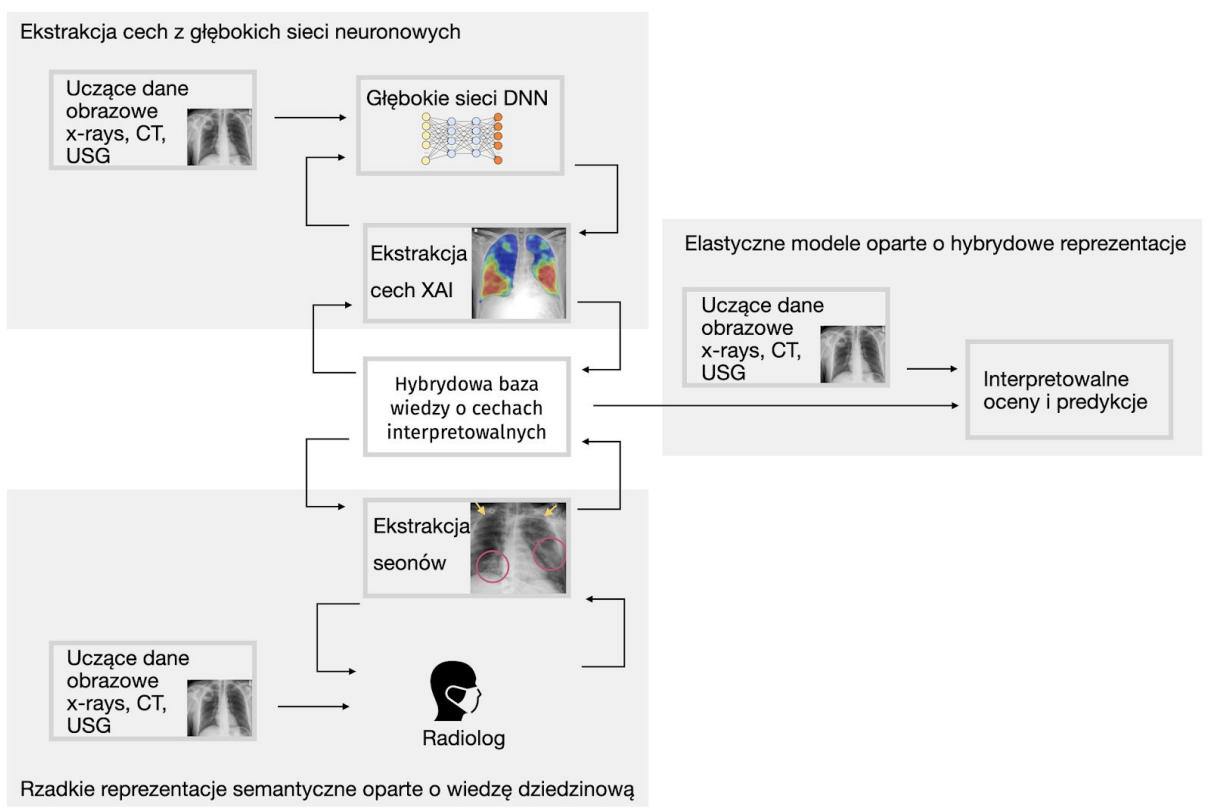
Covid-19 is an infectious respiratory disease. A coronavirus infection leaves permanent ramifications in the respiratory system and beyond. In this situation, tools supporting diagnosis and assessment of lung damage after infection and during Covid-19 treatment are crucial. Preliminary results of analysis of CT images and lung xrays suggest that they can help to quickly assess even asymptomatic cases and facilitate prognosis of response to treatment. There are also reports of usefulness of ultrasound images.
The aim of the DeCoviD project is to develop methods and tools to support radiologists in the assessment of lung imaging data for the occurrence of changes caused by Covid-19 disease. The developed solution will allow to automate the identification of pathological changes and will support the diagnosis of coexisting lung diseases as well as diseases of other organs visible on chest images. It will also allow to quantify the severity of lung damage caused by the disease
Responsible decision support for radiologists requires models based on interpretable features. Such features will be stored in a hybrid knowledge base powered by two research teams from WUT, working on the basis of two, seemingly opposite, paradigms of image data analysis. The eXplainable Artificial Intelligence (XAI) team will use trained deep networks to automatically extract features that are essential for effective disease detection. Cognitive Compressed Sensing (CCS) will build a set of interpretable semantic features using sparse cognitive representations agreed with a group of cooperating radiologists. Combining these two approaches will achieve high effectiveness of the constructed models, combined with high transparency, clarity and stability of the solution.
The DeCoviD project is a part of a broader strategy of competence development in the area of deep learning + XAI + medical applications at the Warsaw University of Technology.
More information: https://github.com/MI2DataLab/DeCoviD.
Work on this project is financially supported by the IDUB against COVID PW.
DALEX 2018-2022
DALEX: Descriptive and model Agnostic Local EXplanations
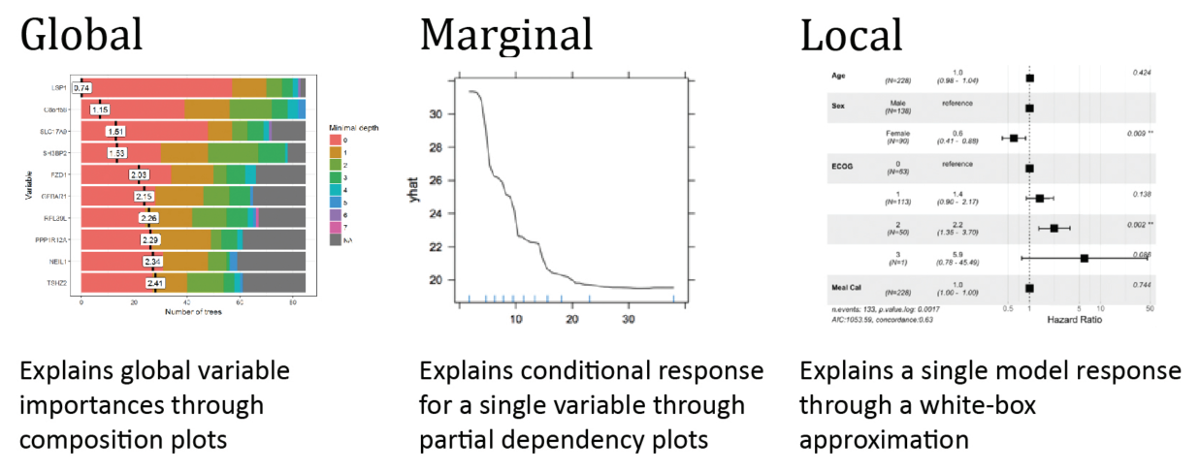
Research project objectives. Black boxes are complex machine learning models, for example deep neural network, an ensemble of trees of high-dimensional regression model. They are commonly used due to they high performance. But how to understand the structure of a black-box, a model in which decision rules are too cryptic for humans? The aim of the project is to create a methodology for such exploration. To address this issue we will develop methods, that: (1) identify key variables that mostly determine a model response, (2) explain a single model response in a compact visual way through local approximations, (3) enrich model diagnostic plots.
Research project methodology. This project is divided into three subprojects - local approximations od complex models (called LIVE), explanations of particular model predictions (called EXPLAIN) and conditional explanations (called CONDA).
Expected impact on the development of science. Explanations of black boxes have fundamental implications for the field of predictive and statistical modelling. The advent of big data forces imposes usage of black boxes that are easily able to overperform classical methods. But the high performance itself does not imply that the model is appropriate. Thus, especially in applications to personalized medicine or some regulated fields, one should scrutinize decision rules incorporated in the model. New methods and tools for exploration of black-box models are useful for quick identification of problems with the model structure and increase the interpretability of a black-box
Work on this project is financially supported from the OPUS grant 2017/27/B/ST6/01307 funded by Polish National Science Centre (NCN).
MLGenSig 2017-2021
MLGenSig: Machine Learning Methods for building of Integrated Genetic Signatures
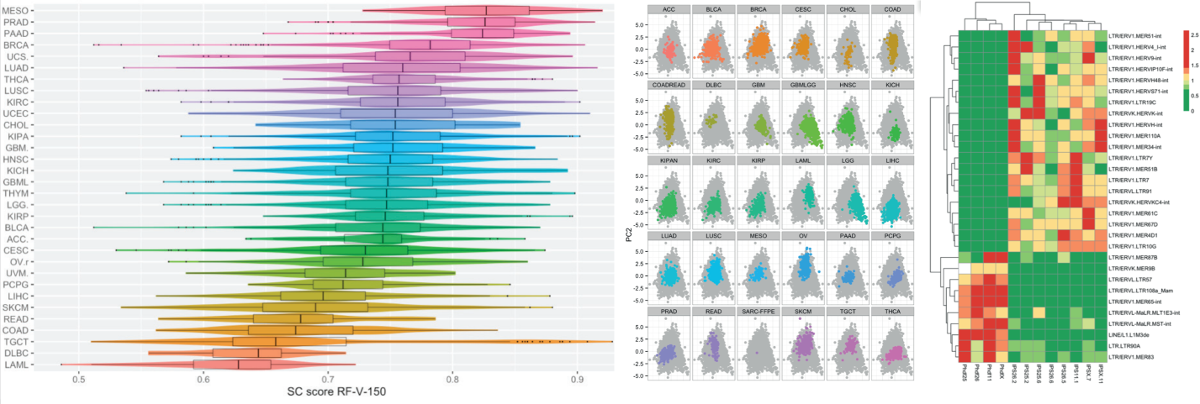
Research project objectives. The main scientific goal of this project is to develop a methodology for integrated genetic signatures based on data from divergent high-throughput techniques used in molecular biology. Integrated signatures base on ensembles of signatures for RNA-seq, DNA-seq, data as well for methylation profiles and protein expression microarrays. The advent of high throughput methods allows to measure dozens of thousands or even millions features on different levels like DNA / RNA / protein. And nowadays in many large scale studies scientists use data from mRNA seq to assess the state of transcriptome, protein microarrays to asses the state of proteome and DNA-seq / bisulfide methylation to assess genome / methylome.
Research methodology. Genetic signatures are widely used in different applications, among others: for assessing genes that differentiate cells that are chemo resistant vs. cells that are not, assess the stage of cell pluripotency, define molecular cancer subtypes. For example, in database Molecular Signatures Database v5.0 one can find thousands of gene sets - genetic signatures for various conditions. There are signatures that characterize some cancer cells, pluripotent cells and other groups. But they usually contain relatively small number of genes (around 100), results with them are hard to replicate and they are collection of features that were found significant when independently tested. In most cases signatures are derived from measurements of the same type. Like signatures based of expression of transcripts based on data from microarrays or RNA-seq, or methylation profile or DNA variation. We are proposing a very different approach. First we are going to use machine-learning techniques to create large collections of signatures. Such signatures base on ensembles of small sub-signatures, are more robust and usually have higher precision. Then out of such signatures we are going to develop methodology for meta-signatures, that integrate information from different types of data (transcriptome, proteome, genome). Great examples of such studies are: Progenitor Cell Biology Consortium (PCBC) and The Cancer Genome Atlas (TCGA) studies. For thousands of patients in different cohorts (for PCBC cohorts based on stemness phenotype, for TCGA based on cancer type) measurements of both mRNA, miRNA, DNA and methylation profiles are available. New, large datasets require new methods that take into account high and dense structure of dependencies between features. The task that we are going to solve is to develop methodology that will create genetic signatures that integrate information from different levels of cell functioning. Then we are going to use data from TCGA and PBCB project to assess the quality of proposed methodology. As a baseline we are going to use following methodologies: DESeq, edgeR (for mRNA), casper (for lternative splicing), MethylKit (for RRBS data) and RPPanalyzer for protein arrays.
Here is the skeleton for our approach: (1) Use ensembles in order to building a genetic signature. The first step would be to use random forests to train a new signature. Ensembles of sub-signtures are build on bootstrap subsamples and they votes if given sample fit given signature or not. (2) In order to improve signatures we are going to consider various normalization of raw counts. We start with log and rank transformation. (3) In order to improve the process of training an ensemble we are going to use pre-filtering of genes. (4) Another approach is to use Bayesian based methods, that may incorporate the expert knowledge, like belief-based gaussian modelling
Research project impact. Genetic profiling is more and more important and has number of application starting from basic classification up to personalized medicine in which patients are profiled against different signatures. Existing tools for genetic signatures have many citations. This we assume that the methodology for integrated genetic profiling will be a very useful for many research groups. It is hard to overestimate the impact of better genetic profiling on medicine. Moreover we build a team of people with knowledge in cancer genetic profiling
Work on this project is financially supported from the OPUS grant 2016/21/B/ST6/02176 funded by Polish National Science Centre (NCN).